Fenilcetonuria
Es una rara afección en la cual un bebé nace sin la capacidad para descomponer apropiadamente un aminoácido llamado fenilalanina.
Causas
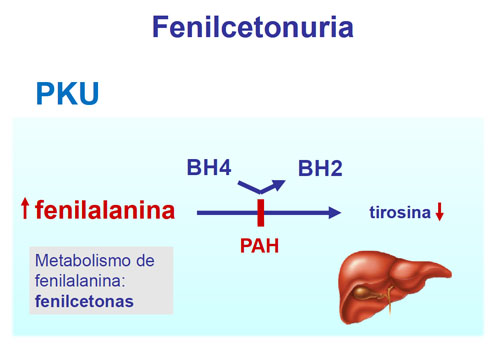
La fenilcetonuria es una enfermedad hereditaria, lo cual significa que se transmite de padres a hijos. Ambos padres deben transmitir una copia defectuosa del gen para que el bebé padezca la enfermedad.
Los bebés con fenilcetonuria carecen de una enzima denominada fenilalanina hidroxilasa. Esta necesaria para descomponer el aminoácido esencial fenilalanina. La fenilalanina se encuentra en alimentos que contienen proteína.
Sin la enzima, los niveles de fenilalanina se acumulan en el cuerpo. Esta acumulación puede dañar el sistema nervioso central y ocasionan daño cerebral.
Síntomas
La fenilalanina juega un papel en la producción corporal de melanina. El pigmento es responsable de dar color a la piel y al cabello. Por lo tanto, los niños con esta afección usualmente tienen un cutis, cabello y ojos más claros que sus hermanos o hermanas sin la enfermedad.
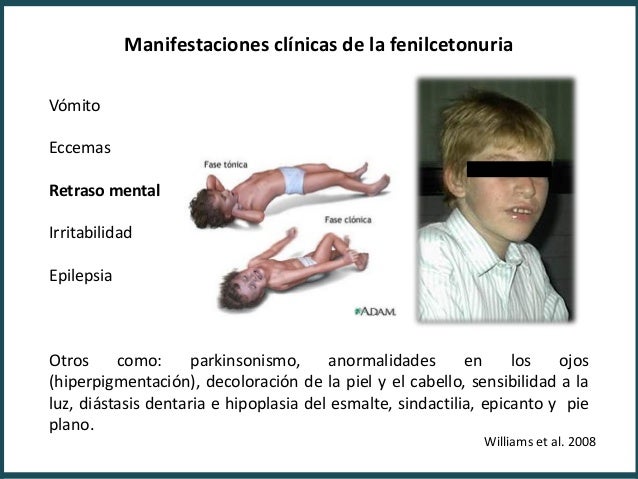
Otros síntomas pueden incluir:
- Retraso de las habilidades mentales y sociales
- Tamaño de la cabeza mucho más pequeño de lo normal
- Hiperactividad
- Movimientos espasmódicos de brazos y piernas
- Discapacidad mental
- Convulsiones
- Erupción cutánea
- Temblores
Si la fenilcetonuria se deja sin tratamiento o si se consumen alimentos que contienen fenilalanina, el aliento, la piel, el cerumen y la orina pueden tener un olor "a ratón" o "a moho". Este olor se debe a la acumulación de sustancias de fenilalanina en el cuerpo.
La fenilcetonuria se puede detectar fácilmente con un simple examen de sangre. Todos los estados de los Estados Unidos exigen una prueba de detección de esta enfermedad para todos los recién nacidos, como parte del grupo de pruebas de detección para neonatos. Este examen generalmente se lleva a cabo tomando unas cuantas gotas de sangre del bebé antes de que este salga del hospital.
Si la prueba de detección es positiva, se requieren exámenes adicionales de sangre y orina para confirmar el diagnóstico. También se llevan a cabo pruebas genéticas
La fenilcetonuria es una enfermedad que se puede tratar. El tratamiento comprende una dieta muy baja en fenilalanina, especialmente cuando el niño está creciendo. La dieta se tiene que seguir en forma estricta. Esto requiere la supervisión exhaustiva por parte del médico o del dietista certificado y la cooperación de los padres y del niño. Quienes que continúan con la dieta hasta la vida adulta tienen una mejor salud física y mental que quienes no lo hacen. Una “dieta de por vida” se ha convertido en la pauta que la mayoría de los expertos recomiendan. Las mujeres que presentan fenilcetonuria deben seguir la dieta antes de la concepción y durante todo el embarazo.
Hay grandes cantidades de fenilalanina en la leche, los huevos y otros alimentos comunes. El edulcorante artificial Nutrasweet (aspartamo) también contiene fenilalanina. Cualquier producto que contenga aspartamo se debe evitar.

Prevención
Un análisis enzimático o una prueba genética pueden determinar si los padres son portadores del gen de la fenilcetonuria (FCU). Asimismo, se puede tomar una muestra de vellosidades coriónicas o amniocentesis durante el embarazo para examinar el feto en búsqueda de esta enfermedad
No hay comentarios:
Publicar un comentario